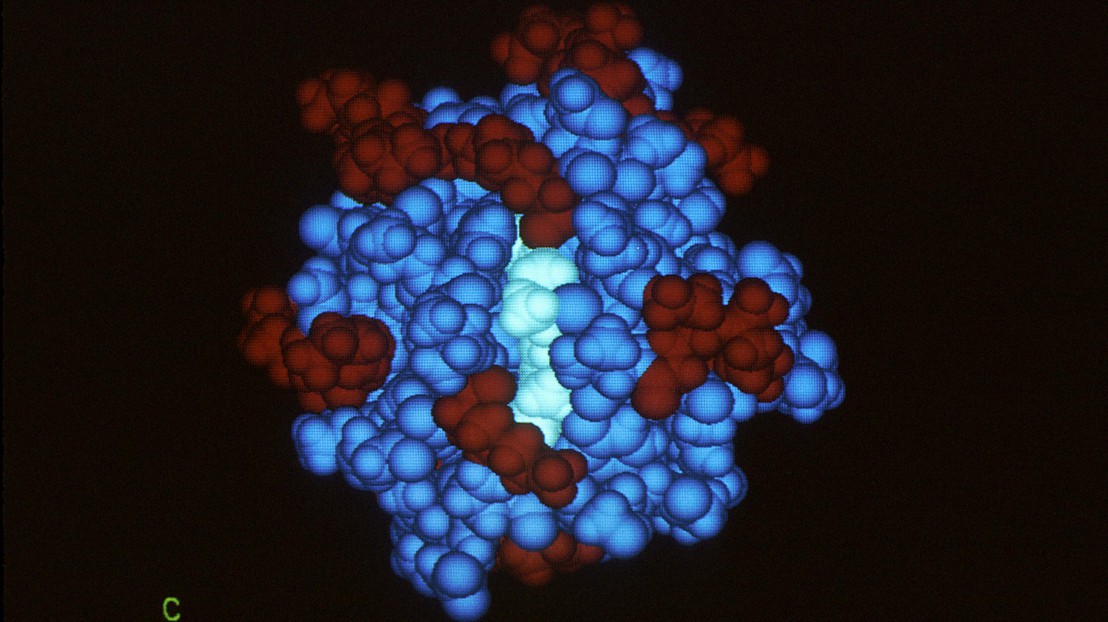
A equipa da Meta AI, a divisão de Inteligência Artificial (IA) da empresa que detém o Facebook, disponibilizou um modelo que é capaz de prever a estrutura de 600 milhões de proteínas. Este novo modelo é baseado em transformadores ESM-2 (um mecanismo que faz uma correlação entre a posição dos elementos em análise, relacionando cada um deles com os que surgem antes e depois) e tem 15 mil milhões de parâmetros. A Meta disponibilizou uma base de dados das suas previsões de estruturas de proteínas, o ESM Metagenomic Atlas. A base de dados inclui também formas de proteínas que ainda não tinham sido observadas pelo cientistas.
As proteínas são moléculas biológicas complexas, que podem ter até 20 tipos de aminoácidos, e são responsáveis por realizar várias funções biológicas nos organismos. Os cientistas precisam de perceber, em toda a plenitude, a forma complexa tridimensional das proteínas para saber como atenuar ou potenciar estas funções biológicas, imitar ou anular esse comportamento. As simulações ou experiências requerem bastante tempo e poder computacional, mas já há modelos de aprendizagem automática (machine learning) que conseguem interpretar a composição química de uma proteína e prever a estrutura, de forma rápida e precisa.
A DeepMind, por exemplo, demonstrou o modelo AlphaFold, que venceu uma competição da especialidade em 2020, e que é capaz de prever a estrutura de mais de 200 milhões de proteínas conhecidas. O modelo ESM da Meta AI, no entanto, vai mais à frente, prevê 600 milhões de formas e foi treinado em outros milhões de sequências de proteínas, noticia o The Register.
Os investigadores explicam que o sistema desenvolvido é “na verdade, um grande modelo de linguagem feito para aprender os padrões evolucionários e gerar previsões de estruturas precisas, de ponta a ponta, diretamente a partir da sequência da proteína”. Os modelos de linguagem moderna contêm dezenas de milhares de milhões de parâmetros e permitem o desenvolvimento de funcionalidades como traduções rápidas, raciocínio de senso comum e resolução de problemas matemáticos, sem influência direta dos programadores (unsupervised learning). A equipa trabalhou com o objetivo de ajustar este modelo para uma solução que consegue prever sequências e formas de proteínas.
O ESM-2 é o maior modelo do género e é capaz de prever as estruturas até 60 vezes mais rápido do que os sistemas anteriores de topo, como o AlphaFold ou o Rosetta, descreve a Meta. A base de dados agora disponibilizada, com 600 milhões de estruturas, demorou duas semanas a ser criada, num sistema com 2000 unidades de processamento gráfico (GPU). Uma única GPU Nvidia V100 demora apenas 14,2 segundos a simular uma proteína constituída por 384 aminoácidos.
A equipa espera que o modelo ESM-2 e a base de dados agora disponibilizados ajudem a comunidade científica a lidar com problemas complexos como combate a doenças ou a alterações climáticas.